Getting Started
Input
To generate profiles you need two input types:
- Reference sequences (FASTA format)
- multi-fasta files are supported
- supported file extensions: .fa, .fasta, .txt
- Short-read sequence data (FASTQ format)
- paired-end and single-end reads are supported
- for paired reads, the last string before the file extension should be ‘_1’ for Read1 and ‘_2’ for Read2 (or ‘_R1’ and ‘_R2’)
- compressed files are supported
- supported file extensions: .fastq, .fq, .fastq.gz, etc.
Generating Profiles
Template for Profile Command
repeatprof profile <'-p' for paired-end reads or '-u' for single-end> <path to reference sequence> <path of the folder containing reads> [optional flags]
Functional Command
repeatprof profile -p Refs.fa /RepeatProfilerData/Test1
Explanation
repeatprofcalls the programprofiledirects the program to generate profiles- see other command options in the documentation
-pindicates the input reads are pairedRefs.faspecifies the path to the FASTA file contianing reference sequences/RepeatProfilerData/Test1specifies the path of the directory containing input read files
Example with Sample Data
Download sample input data set provided here.
- Install RepeatProfiler
- Unzip sample data download and navigate to the folder
- Enter the following command:
repeatprof profile -p reference.fa <enter full path of current directory>
If the program runs without errors, everything should be set up properly.
For more commands and optional flags see the documentation.
Correlation Analysis
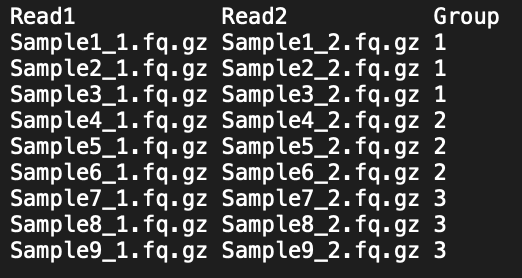
If you are making profiles for multiple samples and want to compare profile shape across samples, you can use the correlation analysis feature (i.e. the -corr flag). This command is designed for cases when multiple samples per category are present (e.g., multiple individuals per species) such that -within group correlation values can be compared to -between group values. The -corr flag requires that you provide an input text file named user_groups.txt that assigns your samples to groups. The user can generate this file manually or use the program to auto-generate the base user_groups.txt file using this command:
repeatprof pre-corr <'-u' for unpaired reads or '-p' for paired reads> <path reads folder>
After running this command, the user_groups.txt will be generated based on your input reads and you can simply replace the placeholder ‘TEMPORARY’ with your group labels such that each sample belonging to a given group has the same label in the ‘group’ column. You can run the following command to view the file and verify that it is in the correct format.
repeatprof pre-corr -v
Example of user_groups.txt